
Overview
Cerebral Cavernous Malformations (CCM) is a blood vessel disorder typified by the development of vascular lesions with the brain and spinal cord. CCM may occur sporadically or as an inherited genetic disease caused by mutation and the resulting loss of function of one of three genes, CCM1, CCM2, or CCM3.
What are the genetics of lesion development?
Genes are packaged in chromosome pairs. We all have two copies of every gene in our genome, with one inherited from each parent. Germline mutations are those that are inherited from a parent, and therefore present in every cell of the body. By contrast, somatic mutations are randomly acquired over time and not present in all cells.
CCM lesions develop when there is a complete loss of function for one of the CCM genes within a brain endothelial (blood vessel) cell. In familial CCM, this means that the affected individual will have inherited a mutation in one of the CCM genes, then at some point in time, acquired a somatic mutation on the other copy of that gene. When this happens in an endothelial cell, there is a complete loss of function for that gene, and lesion development is the result.
There are two forms of cavernous malformation – familial and sporadic. By contrast to the familial, sporadic CCM is not inherited, individuals do not have germline CCM gene mutations, and typically affected individuals present with single lesions. All CCM lesions look like; there is no structural or histological difference between sporadic and familial lesions, indicating that they may develop following the same mechanism. Recent research shows that this is the case – the formation of sporadic lesions is seeded by the acquisition of two somatic mutations. This likely explains why sporadic CCM patients typically develop solitary lesions – it would be exceedingly rare for an individual to acquire two mutations on both copies of the same gene within a single cell.
CCM Lesions
Histologically, at the level of the tissue, all CCM lesions look the same – this similarity is regardless of genotype or sporadic lesions. CCM lesions are composed of dilated vessels, a single layer of endothelium, and they lack the normal supportive structures. Within the lesion tissue, there is an increase in cellular proliferation and angiogenesis, as well as a heightened inflammatory response. Signaling and structural aberrations result in defected junctions, leakiness, bleeding, and iron deposits.
Tumor or Malformations?
Historically, cavernous malformation lesions classify as non-cancerous malformations. A malformation is something that may be present in the body at birth and grows in size through remodeling as the host grows and ages into adulthood. By contrast, a hallmark feature of tumors is growth by a process called clonal expansion. This is where a cell gains some sort of advantage (through mutation, or something else) that causes it to replicate over and over again in an unregulated fashion. The result is a mass of identical cells that can become a tumor.
Advances in imaging technology demonstrate that cavernous malformation lesions are dynamic; we now know that they grow through both remodeling and endothelial cell proliferation and clonal expansion. Therefore, cavernous malformations are somewhat of a hybrid between a malformation and a tumor.
Tumor Mechanism & Biology
Research data shows that cavernous malformations display classical cancer mechanisms. For example, the genetics that underly lesion genesis follow a Knudsonian two-hit model. That means that inactivation (loss of function mutation) of both copies of a gene must occur in order to cause changes that result in the development of the lesion or tumor. In familial CCM, one mutation is inherited, while the other is randomly acquired. This mutational event changes the cell such that it starts to grow by clonal expansion. Mouse research shows that as the lesion matures and becomes multi-cavernous, those mutant cells also recruit non-mutated blood vessel cells into the lesion.
Also, like a tumor, lesion development involves TGF-B mediated endothelial-to-mesenchymal transition. Typically, blood vessels are pretty stable; they don’t change shape or move. Their purpose (especially in the brain) is to hold tight to one another and keep blood inside the vessel. Within a cavernous malformation lesion, however, some of the endothelial cells take on different properties – they move, they become active and express a host of chemical signals that are different from their quiet neighbors.
Researching the biological signaling and genetics associated with the lesion helps us understand better how lesions develop and identify new targets for future therapeutic intervention.
Lesion Structure
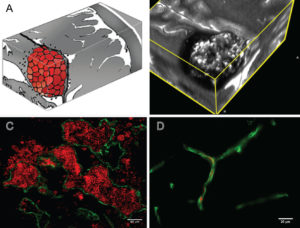

Figure Legend: A: Artist rendition of the mulberry-like CA. B: 3D MRI slab of a human lesion, T2 acquisition at 3 Tesla, highlighting the characteristic “popcorn appearance” of a CA with a hemosiderin ring. C: Confocal immunofluorescence photomicrograph with staining (CD31, green) of endothelial cells (ECs) lining the lesion’s vascular spaces (caverns). Red blood cells (red) fill the caverns and extravasate beyond the “leaky” endothelium. Bar = 40 μM. D: Comparative image of normal brain capillaries. Bar = 20 μM.
The above figure is published in cavernous malformations: deconstructing a neurosurgical disease, an invited review article in the Journal of Neurosurgery, written by Drs. Issam Awad and Sean Polster. The entirety of the article is freely available and recommended for those who would like to learn more about the biology of cavernous malformations, clinical features of the illness, and biomarkers.
To learn more about the drug development pipeline, please visit Treatments in Development.
Reviewed 4.16.22
